
About ‘Omics
three domains of life
-
Eukaryotes - nucleus
-
Bacteria - no nucleus
-
Archaea - no nucleus
the central dogma of biology
DNA —>[transcription]—> mRNA—>[translation]—> polypeptide-chain—>[modification]—>protein
Differences in translation between eukaryotes (human cell) and prokaryotes (bacterium):
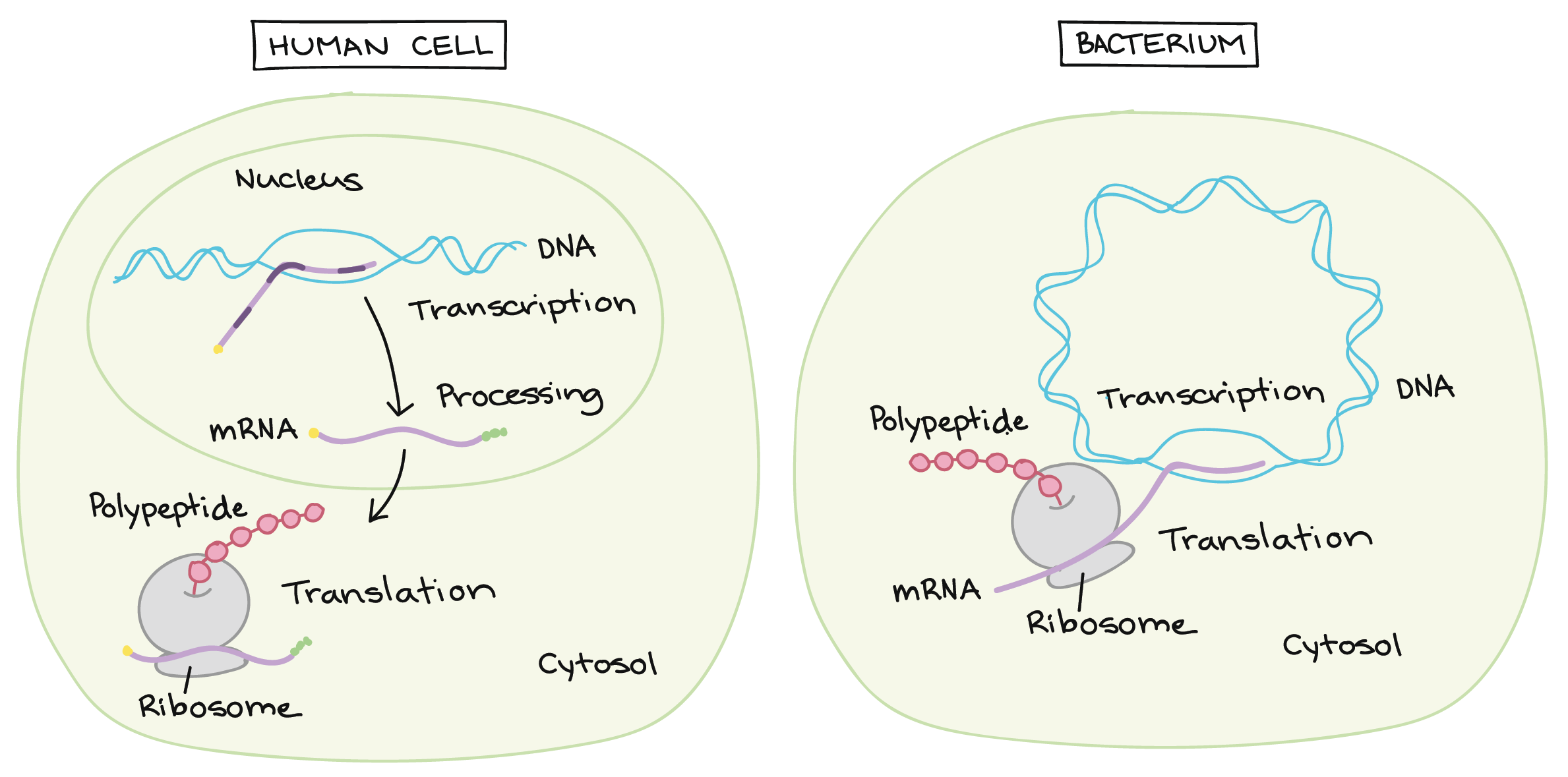 figure source: Khan Academy AP College Biology Unit 6 Lesson 4
figure source: Khan Academy AP College Biology Unit 6 Lesson 4
definitions
| jargon | description |
|---|---|
| bp | Base-pairs (G-C) (A-T) |
| epigenomics | Modifications to DNA that affect gene expression without altering the DNA sequence; primarily DNA methylation and histone modification |
| genome | An organism’s complete set of DNA sequences across all chromosomes, including all genes, and ultimately the three-dimensional form that it takes |
| microbiome | The genomes of bacteria, archaea, and fungi that collectively reside in an environment. The microbiome can be investigated using Shotgun Metagenome Sequencing and 16S rRNA Sequencing. |
| microbiota | The living community of archaea, bacteria, and fungi in an environment |
| transcriptome | An organism’s complete set of RNA, a collection of all the transcript readouts present in a cell |
| Mb / Mbp | Genome size is the total amount of DNA contained within one copy of a single complete genome and can be measured as the total number of nucleotide base pairs, usually in megabases (millions of base pairs, abbreviated Mb or Mbp) |
16s rRNA amplicon sequencing
WHY Choose 16S over Shotgun Metagenomic Sequencing? WATCH THIS ➡️ https://www.youtube.com/watch?v=kXYJ_Dc9qcc
RNA-seq
WATCH THIS ➡️ https://www.youtube.com/watch?v=tlf6wYJrwKY
READ THIS ➡️ Introduction to RNA-seq using high-performance computing, was made by the Harvard Chan Bioinformatics Core github.com/hbctraining 👌🙌.
For even more detail, READ THIS lesson material ➡️ https://scienceparkstudygroup.github.io/rna-seq-lesson/ “Bliek Tijs, Frans van der Kloet and Marc Galland” (eds): “RNA-seq lesson.” Version 2020.04. https://github.com/ScienceParkStudyGroup/rnaseq-lesson
Simply put, RNA-seq (Ribonucleic Acid Sequencing) can quantify gene expression: “which genes are turned on (active) and, how much they are transcribed.”
The transcriptome is defined as a collection of all the transcript readouts present in a cell. RNA-seq data can be used to explore and/or quantify the transcriptome of an organism, which can be utilized for the following types of experiments:
-
Differential Gene Expression: quantitative evaluation and comparison of transcript levels
-
Transcriptome assembly: building the profile of transcribed regions of the genome, a qualitative evaluation.
-
Can be used to help build better gene models, and verify them using the assembly
-
Metatranscriptomics or community transcriptome analysis
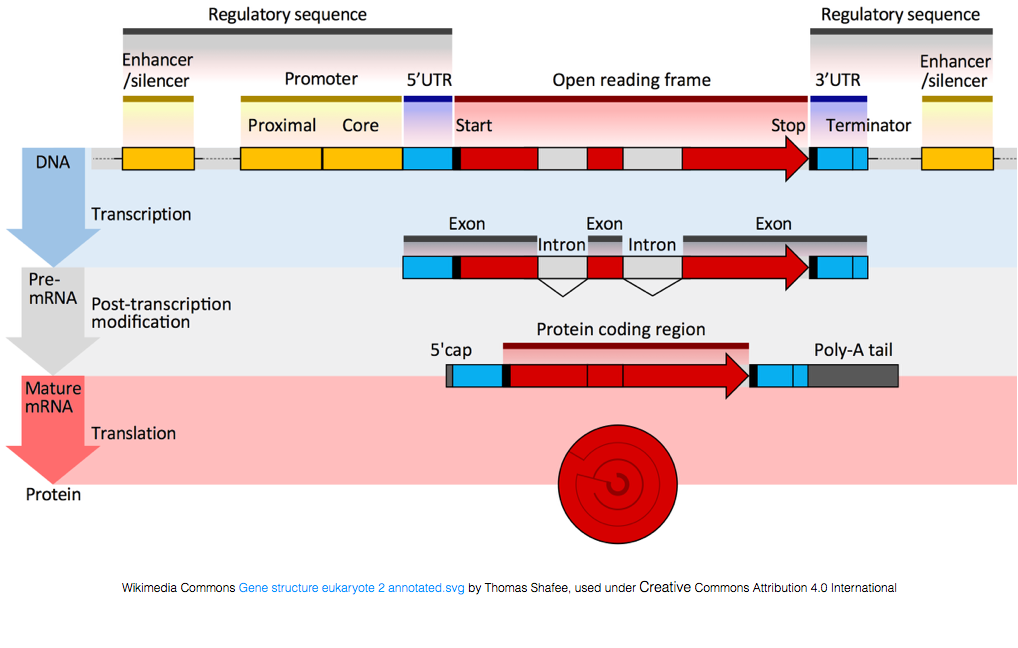
Single Nucleotide Polymorphisms (SNPs)
Read Depth/ Read per Sample
The number of times a particular base is represented within all the reads from sequencing. The higher the read depth, the more confidence scientists can have in identifying a base – known as ‘base calling’. By sequencing each fragment numerous times to produce multiple reads, scientists can be more confident that any variants identified are true variants and not artefacts from the sequencing process. The number of times each individual base has been sequenced i.e. the number of reads it appears in is referred to as the read depth, and the greater the depth, the more confident scientists can be that the variant is real.
*Generally, we recommend 5-10 million read pairs per sample for small genomes (e.g. bacteria) and 20-30 million read pairs per sample for large genomes (e.g. human, mouse). Medium genomes often depend on the project, but 15-20 million read pairs per sample is typically sufficient. For de novo transcriptome assembly projects, we recommend 100 million read pairs per sample.
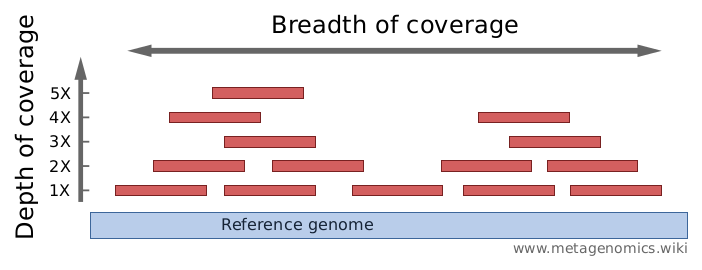 [https://www.metagenomics.wiki/pdf/qc/coverage-read-depth]
[https://www.metagenomics.wiki/pdf/qc/coverage-read-depth]
Total RNA Sample Submission Requirements
| company | min reads per sample | sample type | min RNA amount | recommended RNA amount | Purity (A260/280) | RIN | buffer | |
|---|---|---|---|---|---|---|---|---|
| Azenta (Genewiz) | Total RNA | 500 ng | >2 ug , >50ng/uL | 1.8 - 2.2 | >=6.0 | nuclease-free water | ||
| UW Northwest Genomics Center | ||||||||
| University of Texas | ||||||||
| Mr. DNA Lab | ||||||||
| FYR Diagnostics |
What method is used to remove ribosomal RNA?
Since ribosomal RNA (rRNA) makes up the majority of total RNA, its removal is necessary for efficient sequencing of other RNA species, such as mRNA, long non-coding RNA (lncRNA), and small RNA.
-
For Standard and Strand-Specific RNA-Seq, you can select either poly-A selection or rRNA depletion methods. Poly-A selection is sufficient for studying mRNA in eukaryotes. Analysis of lncRNA or bacterial transcripts requires rRNA depletion.
-
For Ultra-Low Input RNA-Seq, the default is to use poly-A selection. However, if your project requires analysis of lncRNA in addition to mRNA, please make a comment on the quote request form, and we can discuss the available options.
### 1. Illumina library preparation
To do RNA-seq, we have to isolate RNA from each sample and turn it into cDNA (complementary). The cDNA that we make is called the ‘library’. Why is a collection of cDNA a library?
The cDNA libraries can be generated in a way to retain information about which strand of DNA the RNA was transcribed from.
Libraries that retain this information are called stranded libraries, which are now standard with Illumina’s TruSeq stranded RNA-Seq kits. Stranded libraries should not be any more expensive than unstranded, so there is not really any reason not to acquire this additional information.
There are 3 types of cDNA libraries available:
- Forward – reads resemble the gene sequence
- Reverse – reads resemble the complement of the gene sequence (TruSeq)
- Unstranded
To make the cDNA library:
-
isolate mRNA or total RNA
-
remove contaminant DNA
-
either remove rRNA( ribosomal RNA depletion) or select mRNA (polyA selection), resulting in fragmented RNA
For differential gene expression analysis, it is best to enrich for Poly(A)+
-
reverse transcribe the fragmented RNA into double-stranded cDNA
-
Add (ligate) sequencing adaptors
Allows sequencing machine to recognize the fragments and to sequence many samples at the same time, different samples can use different adaptors
-
PCR amplify the library
Only the fragments with sequencing adaptors are amplified
-
fragments are size selected (usually 300-500bp)
-
Quality Control
### 2. Illumina Sequencing
Fragments are laid out vertically in a grid, called a flow cell. Fluorescent probes
Low quality score
-
Low confidence : fluorescent probe didn’t shine bright enough
-
Low diversity : overabundance of a single color making it hard to identify individual sequences (the colors blue together) especially problematic in the first few nucleotidesof a sequence because that is when the sequencer determines where the DNA fragments are located on the flow cell
Each sequencing read has 4 lines of data:
1ST LINE: Unique ID for sequence
2ND LINE: Bases for sequenced fragment ex. (ACAGACGGATGACGA)
3RD LINE: ‘+’ spacer character
4TH LINE: quality scores for each base in the sequenced fragments
Filter out garbage reads
- reads with low quality base calls
- reads that are adaptor sequences bound to each other (no DNA fragment)
Align high quality reads to a genome
- Index of all fragments and locations in the genome
Count the number of reads per gene
- ‘Bulk’ RNA-seq sample is the average of a pool of the same treatment
Normalize the data
- low quality reads
- how many reads were successful
3. Data Analysis
The data is a huge matrix.
Principal component analysis (PCA)
Marine Omics Fitting Multifactorial Models of Differential Expression, RNA-seq
